A hypercoagulable disorder, also known as thrombophilia, is an inherited or acquired condition that increases the risk of developing inappropriate or excessive thrombus (blood clot) formation. In general within the population acquired hypercoagulable disorders are more common than inherited disorders.
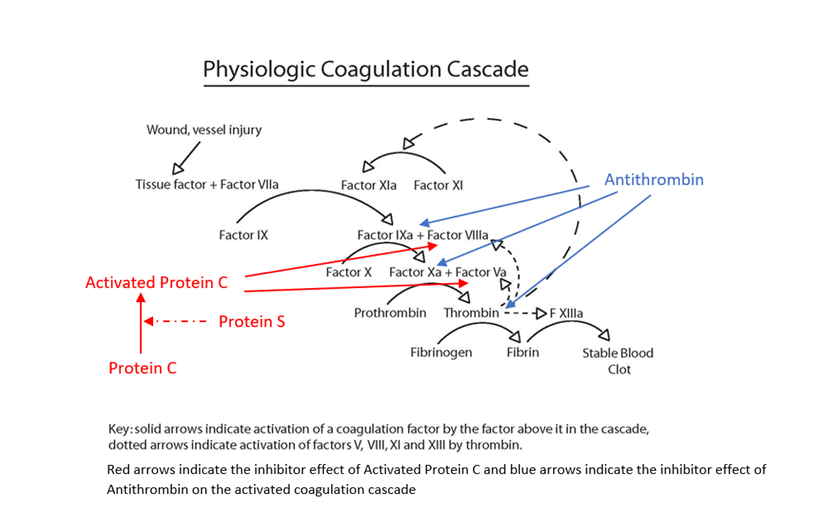
Blood clotting is normal response to blood vessel or tissue injury. Blood is normally in a fluid state (anticoagulated) within the body to enable flowing through a vessel network of arteries, capillaries and veins, delivering oxygen and nutrients to the organs of the body and removing carbon dioxide and waste products. When a blood vessel is injured, it begins to leak blood, either externally e.g a skin cut or internally into body tissues e.g a bruise. The body stops this blood loss through a complex clotting process called haemostasis. During haemostasis, the injured blood vessel becomes narrower (constricts) to reduce blood flow, cells in the blood called platelets stick (adhere) to the injury site and clump together to form a loose platelet plug and the coagulation cascade is triggered (initiated). During the cascade process, the body sequentially activates coagulation factors, which are proteins that create a net of fibrin threads, which weave them through the platelet plug making a stable firm fibrin blood clot, whilst still allowing blood to flow through the damage vessel. This blood clot covers the injured area and forms a barrier to prevent further blood loss and the clot should stay in place until the injury has healed.
In normal haemostasis once the body has activated the clotting process, there are regulatory feedback mechanisms, which limit and control the clotting process (natural anticoagulants), prevent the complete blocking of the vessel by excessive clot formation and by removal of the fibrin clot once the injury has healed (fibrinolysis). Hypercoagulable disorders can occur when something goes wrong within this clotting process. If the clotting process activates inappropriately, or feedback mechanisms fail to work effectively to limit formation or removal of fibrin clot, then there can be inappropriate and/or excessive blood clot formation.
Presentation of Hypercoagulable Disorders
Thrombosis can occur within veins or arteries, however the mechanism of clot formation is different, with venous thrombosis associated with sluggish movement of blood (stasis) or imbalance of the clotting progress and feedback mechanism, whereas arterial thrombosis more commonly results from the rupture of an atherosclerotic plaque due to build-up of cholesterol in the arterial wall. Blood clots are referred to as thrombi (one = thrombus) when they form in a blood vessel; thrombi may break off and block another blood vessel in another part of the body, where they are referred to as emboli (one =embolus).
- Venous thromboembolism (VTE) is the most common condition associated with hypercoagulable disorders, with blood clots most frequently forming in the deep veins of the legs (DVT) causing redness, pain and swelling particularly at the back of legs. However, a DVT can become life-threatening if the clot breaks free and travels to other parts of the body through the bloodstream, particularly if it becomes lodged in the arteries of lung, called a pulmonary embolism PE or to the brain causing strokes.
- Although less common, thrombosis can occur in unusual venous circulation sites causing cerebral vein thrombosis (brain), hepatic and portal vein thrombosis (liver), mesenteric vein thrombosis (small intestines), renal vein thrombosis (kidney), venous thrombosis in arms and ovarian vein thrombosis
- Recurrent foetal loss (miscarriages) and other complications in pregnancy may be associated with thrombophilia.
- Pupura fulminas is the result of severe (homozygous) Protein C or Protein S deficiency causing massive thromboembolic complications (a form disseminated intravascular coagulation) in newborns shortly after birth. It is life-threatening causing tissue death (necrosis) and bleeding under the skin and other organs, without treatment with Protein C concentrate or fresh frozen plasma. It can also occur in adults associated with acquired rapid onset following acute infections.
- Warfarin medication may also produce a similar phenomenon to purpura fulminas during the early days of starting therapy due to reducing the level of Protein C which has a short half-life, before significant falls in the other vitamin K dependent procoagulants occurs.
- Disseminated intravascular coagulation (DIC) is a life-threatening, acute, acquired condition that causes tiny clots throughout the body,often associated with sepsis. It uses up coagulation factors and platelets at an accelerated rate, leading to both formation of thrombi in many organs and bleeding.
- Immunothrombosis is one of the significant complications of severe COVID-19 infections is a coagulopathy that seems to be related to the occurrence of venous and arterial thromboembolic disease, with strong local pulmonary thrombotic microangiopathy and direct endothelial cell infection and injury by the virus. The coagulation changes mimic but are not identical and do not meet the criteria for disseminated intravascular coagulation.
Risk factors for Hypercoagulable Disorders
Most people have the potential to develop a blood clot but there are several situations or conditions that can increase an individual’s risk of a hypercoagulable disorder including:
- Inherited condition that increases the risk of thrombosis or family history of thrombosis
- Decreased blood flow due to prolonged periods of immobility
- Admission to hospital
- Injury or surgery affecting a vein
- Increased oestrogen due to pregnancy or hormone therapy
- Medical conditions linked with cancer, inflammatory disorders, heart failure, atrial fibrillation, hypertension, antiphospholipid syndrome and obesity
- Increasing age
Certain inherited gene mutations that may predispose someone to hypercoagulable states, such as factor V Leiden or the prothrombin G20210 A mutation, are relatively common in the population, but it is thought that they add only a slight increase in the risk of actually developing a problem with clotting. Other inherited hypercoagulable disorders, such as protein C deficiency, protein S deficiency, and antithrombin deficiency have a higher risk of thrombosis but are relatively rare. These are generally due to genetic mutations that lead to a deficiency or dysfunction in the coagulation protein that the gene produces. All of the inherited disorders (except for antithrombin deficiency) may be seen in heterozygous (one gene copy) or homozygous (two gene copies) form. If someone has two mutated gene copies, they tend to have a more severe form of the condition, and if they are heterozygous in more than one condition, the risk of clotting tends to be additive (and sometimes they multiply the risk). With inherited hypercoagulable disorders, the first thrombotic episode may be seen at a relatively young age (less than 40 years of age).